INDRA
![]()
![]()
![]()

![]()
INDRA (Integrated Network and Dynamical Reasoning Assembler) is an automated model assembly system, originally developed for molecular systems biology and then generalized to other domains (see INDRA World). INDRA draws on natural language processing systems and structured databases to collect mechanistic and causal assertions, represents them in a standardized form (INDRA Statements), and assembles them into various modeling formalisms including causal graphs and dynamical models.
At the core of INDRA are its knowledge-level assembly procedures, allowing sources to be assembled into coherent models, a process that involves correcting systematic input errors, finding and resolving redundancies, inferring missing information, filtering to a relevant scope and assessing the reliability of causal information.
The detailed INDRA documentation is available at http://indra.readthedocs.io.
Contents
INDRA Modules
Knowledge sources
INDRA is currently integrated with the following natural language processing
systems and structured databases. These input modules (available in
indra.sources) all produce INDRA Statements.
Reading systems:
| Reader | Module | Reference |
|---|---|---|
| TRIPS/DRUM | indra.sources.trips |
http://trips.ihmc.us/parser/cgi/drum |
| REACH | indra.sources.reach |
https://github.com/clulab/reach |
| Sparser | indra.sources.sparser |
https://github.com/ddmcdonald/sparser |
| Eidos | indra.sources.eidos |
https://github.com/clulab/eidos |
| TEES | indra.sources.tees |
https://github.com/jbjorne/TEES |
| MedScan | indra.sources.medscan |
https://doi.org/10.1093/bioinformatics/btg207 |
| RLIMS-P | indra.sources.rlimsp |
https://research.bioinformatics.udel.edu/rlimsp |
| ISI/AMR | indra.sources.isi |
https://github.com/sgarg87/big_mech_isi_gg |
| Geneways | indra.sources.geneways |
https://www.ncbi.nlm.nih.gov/pubmed/15016385 |
| GNBR | indra.sources.gnbr |
https://zenodo.org/record/3459420 |
| SemRep | indra.sources.semrep |
https://github.com/lhncbc/SemRep |
Biological pathway databases:
| Database / Exchange format | Module | Reference | |
|---|---|---|---|
| PathwayCommons / BioPax | indra.sources.biopax |
http://pathwaycommons.org/ http://www.biopax.org/ |
|
| Large Corpus / BEL | indra.sources.bel |
https://github.com/pybel/pybel https://github.com/OpenBEL |
|
| Signor | indra.sources.signor |
https://signor.uniroma2.it/ | |
| BioGRID | indra.sources.biogrid |
https://thebiogrid.org/ | |
| Target Affinity Spectrum | indra.sources.tas |
https://doi.org/10.1101/358978 | |
| HPRD | indra.sources.hprd |
http://www.hprd.org | |
| TRRUST | indra.sources.trrust |
https://www.grnpedia.org/trrust/ | |
| Phospho.ELM | indra.sources.phosphoelm |
http://phospho.elm.eu.org/ | |
| VirHostNet | indra.sources.virhostnet |
http://virhostnet.prabi.fr/ | |
| CTD | indra.sources.ctd |
http://ctdbase.org | |
| DrugBank | indra.sources.drugbank |
https://www.drugbank.ca/ | |
| OmniPath | indra.sources.omnipath |
https://omnipathdb.org/ | |
| DGI | indra.sources.dgi |
https://www.dgidb.org/ | |
| CRoG | indra.sources.crog |
https://github.com/chemical-roles/chemical-roles | |
| CREEDS | indra.sources.creeds |
https://maayanlab.cloud/CREEDS/ | |
| UbiBrowser | indra.sources.ubibrowser |
http://ubibrowser.ncpsb.org.cn/ | |
| ACSN | indra.sources.acsn |
https://acsn.curie.fr/ACSN2/ACSN2.html | |
| WormBase | indra.sources.wormbase |
https://www.wormbase.org |
Custom knowledge bases:
| Database / Exchange format | Module | Reference |
|---|---|---|
| NDEx / CX | indra.sources.ndex_cx |
http://ndexbio.org |
| INDRA DB / INDRA Statements | indra.sources.indra_db_rest |
https://github.com/indralab/indra_db |
| Hypothes.is | indra.sources.hypothesis |
https://hypothes.is |
| Biofactoid | indra.sources.biofactoid |
https://biofactoid.org/ |
| MINERVA | indra.sources.minerva |
https://covid19map.elixir-luxembourg.org/minerva/ |
Output model assemblers
INDRA also provides several model output assemblers that take INDRA Statements as input. The most sophisticated model assembler is the PySB Assembler, which implements a policy-guided automated assembly procedure of a rule-based executable model (that can then be further compiled into other formats such as SBML, Kappa, BNGL and SBGN to connect to a vast ecosystem of downstream tools). Several other model assembly modules target various network formats for visualization, and graph/structural analysis (PyBEL, CyJS, Graphviz, SBGN, CX, SIF, etc.) and curation (HTML, TSV, IndexCards). Finally, the English Assembler produces English language descriptions of a set of INDRA Statements.
INDRA also supports extension by outside model assembly tools which take INDRA Statements as input and produce models. One such example is Delphi (https://github.com/ml4ai/delphi), which is a Dynamic Bayesian Network model assembler. Similarly, outside tools that support INDRA Statements can implement custom visualization methods, such as CauseMos, developed by Uncharted Software (https://uncharted.software/).
Assemblers aimed at model-driven discovery and analysis:
| Modeling formalism / Exchange format | Purpose | Module | Reference |
|---|---|---|---|
| PySB (-> SBML, SBGN, BNGL, Kappa, etc.) | Detailed, mechanistic modeling, simulation, analysis | indra.assemblers.pysb |
http://pysb.org |
| PyBEL | Causal analysis, visualization | indra.assemblers.pybel |
https://github.com/pybel/pybel https://bel-commons.scai.fraunhofer.de/ |
| IndraNet | Causal analysis, signed and unsigned | indra.assemblers.indranet |
|
| SIF | Network analysis, logic modeling, visualization | indra.assemblers.sif |
SIF format |
| KAMI | Knowledge aggregation of protein sites/states and Kappa modeling | indra.assemblers.kami |
https://github.com/Kappa-Dev/KAMI |
Assemblers primarily aimed at visualization:
| Network / Exchange format | Purpose | Module | Reference |
|---|---|---|---|
| Causal Analysis Graph | General causal graph visualization | indra.assemblers.cag |
|
| CX | Network browsing, versioning on NDEx | indra.assemblers.cx |
http://ndexbio.org |
| Cytoscape JS | Interactive Cytoscape JS network to embed in websites | indra.assemblers.cyjs |
http://js.cytoscape.org/ |
| Graphviz | Static PDF/PNG visualization with powerful automated layout using Graphviz | indra.assemblers.graph |
https://www.graphviz.org/ |
| SBGN | Visualization with Systems Biology Graphical Notation | indra.assemblers.sbgn |
http://sbgn.org |
Assemblers primarily aimed at expert curation and browsing:
| Output format | Purpose | Module | Reference |
|---|---|---|---|
| English language | Human-readable descriptions, reports, dialogue | indra.assemblers.english |
|
| HTML | Web-based browsing, linking out to provenance, curation | indra.assemblers.html |
Curation tutorial |
| TSV (Tab/Comma Separated Values) | Spreadsheet-based browsing and curation | indra.assemblers.tsv |
|
| Index Cards | Custom JSON format for curating biological mechanisms | indra.assemblers.index_card |
Internal knowledge assembly
A key feature of INDRA is providing internal knowledge-assembly modules that operate on INDRA Statements and perform the following tasks:
- Redundancy/subsumption/generalization/contradiction finding and resolution
with respect to an ontology with the Preassembler
(
indra.preassembler.Preassembler) - Belief calculation based on evidence using the BeliefEngine
(
indra.belief) - Mapping grounding between multiple ontologies
(
indra.preassembler.ont_mapper.OntMapper) - Grounding override and disambiguation
(
indra.preassembler.grounding_mapper.GroundingMapper) - Protein sequence mapping (
indra.preassembler.site_mapper.SiteMapper)
The internal assembly steps of INDRA including the ones listed above, and also a large collection of filters (filter by source, belief, the presence of grounding information, semantic filters by entity role, etc.) are exposed in the indra.tools.assemble_corpus submodule. This submodule contains functions that take Statements as input and produce processed Statements as output. They can be composed to form an assembly pipeline connecting knowledge collected from sources with an output model.
This diagram illustrates the assembly pipeline process.
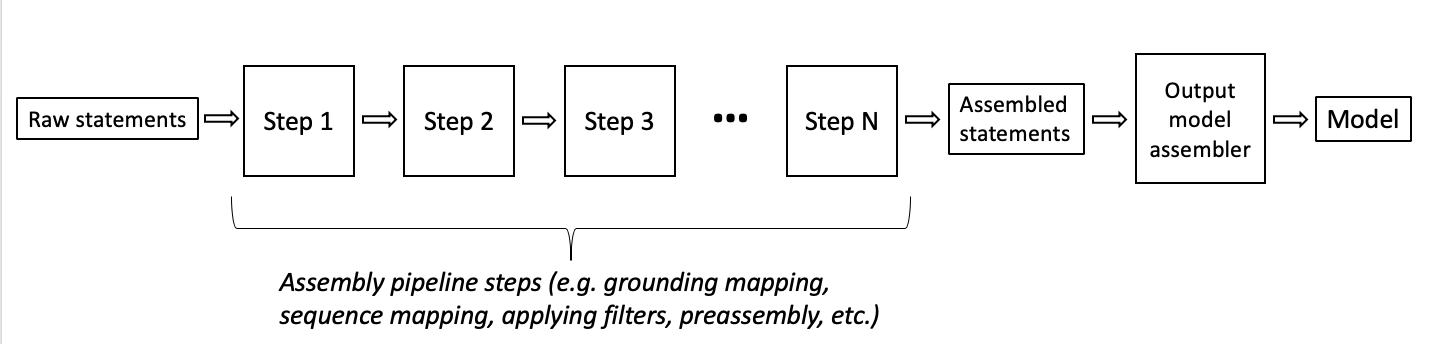
The choice of assembly functions can vary depending on the domain (i.e, biology or world modeling), the modeling goal (i.e., the type of model that will be assembled and how that model will be used), desired features, and confidence (e.g., filter to human genes only or apply a belief cutoff), and any other user preferences.
An example of a typical assembly pipeline for biology statements is as follows. Some of the below steps can be removed, rearranged, and other steps added to change the assembly pipeline.
from indra.tools import assemble_corpus as ac
stmts = <the collection of all raw statements to use>
stmts = ac.filter_no_hypothesis(stmts) # Filter out hypothetical statements
stmts = ac.map_grounding(stmts) # Map grounding
stmts = ac.filter_grounded_only(stmts) # Filter out ungrounded agents
stmts = ac.filter_human_only(stmts) # Filter out non-human genes
stmts = ac.map_sequence(stmts) # Map sequence
stmts = ac.run_preassembly(stmts, # Run preassembly
return_toplevel=False)
stmts = ac.filter_belief(stmts, 0.8) # Apply belief cutoff of 0.8
An example of an assembly pipeline for statements in the world modeling domain
is as follows (note how biology-specific functions are not used, and a custom
belief_scorer and ontology is passed to run_preassembly here, while the
biology pipeline used default values). Note that this example requires
the indra_world package to be installed.
from indra.tools import assemble_corpus as ac
from indra_world.belief.wm_scorer import get_eidos_scorer
from indra_world.ontology.world import world_ontology
stmts = <the collection of all raw statements to use>
stmts = ac.filter_grounded_only(stmts) # Filter out ungrounded agents
belief_scorer = get_eidos_scorer()
stmts = ac.run_preassembly(stmts, # Run preassembly
return_toplevel=False,
belief_scorer=belief_scorer,
ontology=world_ontology,
normalize_equivalences=True, # Optional: rewrite equivalent groundings to one standard
normalize_opposites=True, # Optional: rewrite opposite groundings to one standard
normalize_ns='WM') # Use 'WM' namespace to normalize equivalences and opposites
stmts = ac.filter_belief(stmts, 0.8) # Apply belief cutoff of e.g., 0.8
Assembled statements returned after running the assembly pipeline can be passed into any of the output model assemblers.
Other modules
INDRA also contains modules to access literature content (e.g., PubMed, Elsevier), available in indra.literature, and
access ontological information and convert between identifiers (e.g., UniProt,
HGNC), available in indra.databases.
A full list of further INDRA modules is available in the documentation.
Citation
Gyori B.M., Bachman J.A., Subramanian K., Muhlich J.L., Galescu L., Sorger P.K. From word models to executable models of signaling networks using automated assembly (2017), Molecular Systems Biology, 13, 954.
Bachman J.A., Gyori B.M., Sorger P.K. Automated assembly of molecular mechanisms at scale from text mining and curated databases (2023), Molecular Systems Biology, e11325.
Installation
For detailed installation instructions, see the documentation.
INDRA currently supports Python 3.8-3.10. The last release of INDRA compatible with Python 2.7 is 1.10, the last release fully compatible with Python 3.5 is 1.17. Most usages of INDRA will work with other Python versions, however, full compatibility is currently only tested with 3.8-3.10.
The preferred way to install INDRA is by pointing pip to the source repository as
$ pip install git+https://github.com/sorgerlab/indra.git
Releases of INDRA are also available on PyPI, you can install the latest release as
$ pip install indra
However, releases will usually be behind the latest code available in this repository.
INDRA depends on a few standard Python packages. These packages are installed by pip during setup. For certain modules and use cases, other “extra” dependencies may be needed, which are described in detail in the documentation.
INDRA REST API
A REST API for INDRA is available at http://api.indra.bio:8000. Note that the REST API is ideal for prototyping and for building light-weight web apps, but should not be used for large reading and assembly workflows.
INDRA Docker
INDRA is available as a Docker image on Dockerhub and can be pulled as
docker pull labsyspharm/indra
You can run the INDRA REST API using the container as
docker run -id -p 8080:8080 --entrypoint python labsyspharm/indra /sw/indra/rest_api/api.py
The Dockerfile to build the image locally is available in the docker folder in this repository.
Using INDRA
In this example INDRA assembles a PySB model from the natural language description of a mechanism via the TRIPS reading web service.
from indra.sources import trips
from indra.assemblers.pysb import PysbAssembler
pa = PysbAssembler()
# Process a natural language description of a mechanism
trips_processor = trips.process_text('MEK2 phosphorylates ERK1 at Thr-202 and Tyr-204')
# Collect extracted mechanisms in PysbAssembler
pa.add_statements(trips_processor.statements)
# Assemble the model
model = pa.make_model(policies='two_step')
INDRA also provides an interface for the
REACH natural language
processor. In this example, a full paper from PubMed
Central is processed. The paper’s PMC ID is
PMC8511698. The example
assumest that a REACH server is running locally (see documentation at
indra.sources.reach).
Note that REACH takes about 8 minutes to read this full-text paper.
from indra.sources import reach
reach_processor = reach.process_pmc('PMC8511698', url=reach.local_nxml_url)
At this point, reach_processor.statements contains a list of INDRA statements
extracted from the PMC paper.
Next we look at an example of reading the 10 most recent PubMed abstracts on BRAF and collecting the results in INDRA statements.
from indra.sources import reach
from indra.literature import pubmed_client
# Search for 10 most recent abstracts in PubMed on 'BRAF'
pmids = pubmed_client.get_ids('BRAF', retmax=10)
all_statements = []
for pmid in pmids:
abs = pubmed_client.get_abstract(pmid)
if abs is not None:
reach_processor = reach.process_text(abs, url=reach.local_text_url)
if reach_processor is not None:
all_statements += reach_processor.statements
At this point, the all_statements list contains all the statements
extracted from the 10 abstracts.
The next example shows querying the BEL large corpus network for a neighborhood of a given list of proteins using their HGNC gene names. This example performs the query via PyBEL.
from indra.sources import bel
# Process the neighborhood of BRAF and MAP2K1
bel_processor = bel.process_pybel_neighborhood(['BRAF', 'MAP2K1'])
At this point, bel_processor.statements contains a list of INDRA statements
extracted from the neighborhood query.
Next, we look at an example of querying the Pathway Commons database for paths between two lists of proteins.
from indra.sources import biopax
# Process the neighborhood of BRAF and MAP2K1
biopax_processor = biopax.process_pc_pathsfromto(['BRAF', 'RAF1'], ['MAP2K1', 'MAP2K2'])
At this point, biopax_processor.statements contains a list of INDRA
Statements extracted from the paths-from-to query.
Funding
The development of INDRA has been funded from the following sources:
| Program | Grant number |
|---|---|
| DARPA Big Mechanism | W911NF-14-1-0397 |
| DARPA World Modelers | W911NF-18-1-0014 |
| DARPA Communicating with Computers | W911NF-15-1-0544 |
| DARPA Automated Scientific Discovery Framework | W911NF-18-1-0124 |
| DARPA Automating Scientific Knowledge Extraction | HR00111990009 |
| DARPA Panacea | HR00111920022 |
| DARPA Young Faculty Award | W911NF-20-1-0255 |
| DARPA ASKEM / ARPA-H BDF | HR00112220036 |